参考案例
基于16S rDNA测序评估种水平和菌株水平的微生物组分析(IF-11.878)
使用计算机模拟和高通量测序实验评估16S rDNA测序在物种和菌株水平上分类的能力。研究结果证明,全长16S测序较短读长的16S测序,有更高的物种分辨率;同时,全长16S测序可以准确的检测SNP。该研究显示,全长16S rDNA测序可以鉴定到“种”和“菌株”水平。
材料和方法
1. 从Greengenes数据库下载全长16S rRNA非冗余基因集,计算机模拟不同可变区。
2. 36种细菌构建模拟微生物群落,使用PacBio CCS模式进行全长16S测序。
3. 4个健康人的粪便样品,分别使用PacBio CCS模式进行全长16S测序、Illumina对V1-V3区测序和宏基因组测序。
4. 人类肠道微生物分离物,使用PacBio CCS模式进行全长16S测序。
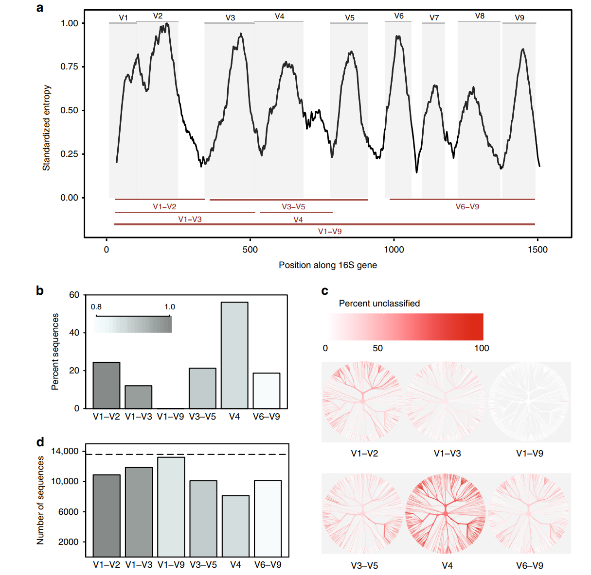
图1 计算机模拟16S rDNA可变区的比较
研究结果
全长16S rDNA测序分类分辨率更好
全长16S rDNA长度约1500bp,包含9个可变区。对全长16S rRNA非冗余基因集序列中包含的PCR引物结合位点剪切后,产生不同可变区的模拟扩增子。
结果发现,不同可变区的物种分类结果存在很大差异。其中V4区表现最差,56%未匹配到物种;V1-V2区对蛋白菌门的鉴定表现不佳;而V3-V5区对放线菌门的鉴定表现不佳;V1-V9区结果最好;当使用全长序列时,几乎所有的模拟扩增子可以匹配到物种。不同亚区的选择显著影响聚类产生OUT的数量,序列相似度达到99%时,所有可变区都未能重现原始数据库中不同序列的数量。
模拟扩增子研究表明,使用短序列对不同亚区进行测序,无法区分相似度高的类群。
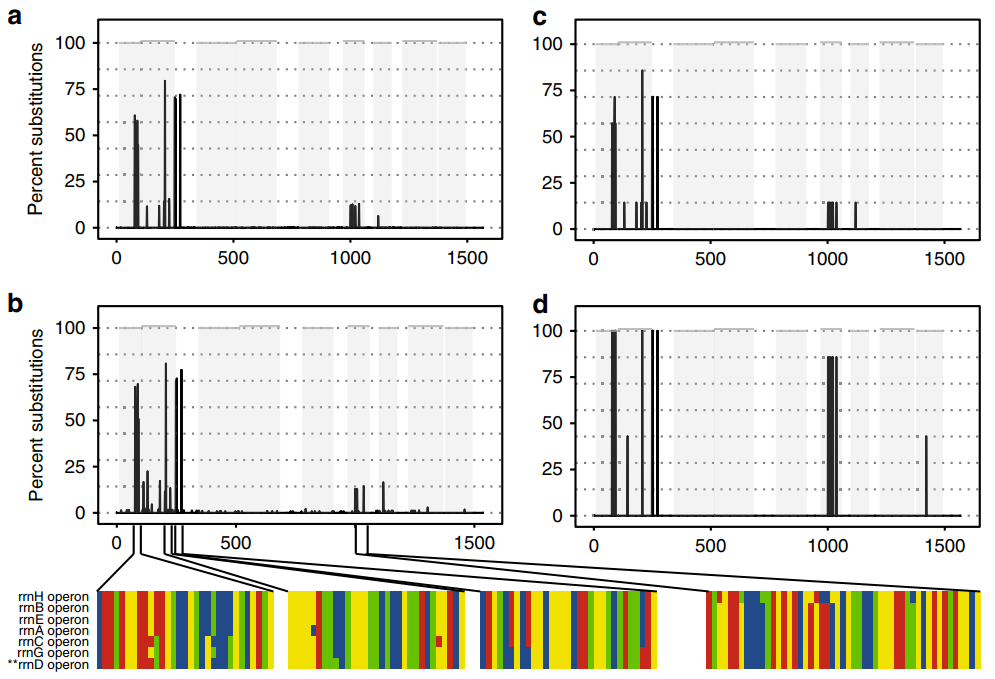
图2 大肠杆菌 16S rDNA序列中的多态性
16S rDNA拷贝数变异反映菌株水平变异
使用36种细菌模拟群体实验,对混合样品进行PacBio CCS模式测序。将测序获得的全长16S序列比对到数据库。发现同一物种内不同16S基因拷贝数之间存在差异,因此不同16S基因拷贝数可以作为鉴别菌株的工具。

图3 检测人类粪便样品中的拟杆菌
复杂微生物组内的16S多态性
选择4个健康人的粪便样品,分别使用全长16S测序、Illumina对V1-V3区测序、同时使用短序列对宏基因组进行测序和分析。结果显示,全长16S和V1-V3区在Bacteroides属的相对丰度相似,但是V1-V3区不能检测到Bacteroides indestinalis。通过对Bacteroides vulgatus分析全长16S rDNA不同拷贝之间的变异检测,显示测序数据中同一个OUT存在碱基替换现象,而且替换特征与已测序的菌株各不相同。表明全长16S rDNA可以分辨非常相近的细菌分类。基因组内16S多态性普遍存在
通过对人类肠道微生物分离物进行全长16S测序,发现这些基因普遍存在多态性;而且,通过比较相同OTU内的SNP特征,发现3种细菌分离株之间存在不同的SNPs频率(图4b-d)。研究表明相似物种之间在基因组内16S基因拷贝变异的差异,可用于分辨相同物种内的菌株。

图4 肠道16S rDNA的多态性
参考文献
Johnson, J.S., Spakowicz, D.J., Hong, B. et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun 10, 5029 (2019).