- Product introduction
- Common problem
- Classic case
- Result display
- Single-cell full-length transcriptome sequencing
Single-cell full-length transcriptome sequencing refers to the use of Nanopore sequencing technology to directly read the reverse-transcribed full-length cDNA of single cells treated with 10X Genomics, identify and quantify the Isoform of each cell by analysis, and detect the single-cell level Alternative Splicing event. Single-cell full-length transcriptome sequencing improves single-cell transcriptome analysis from the gene level to the isoform level by combining the advantages of single cells and three-generation long-read technology.
Product advantages
New isoform of cell group specificity
Different cell groups have different variable shear
Differentially expressed transcripts in different cell groups
Full length transcripts of cell groups
Fusion gene of cell groups
Sequencing service process
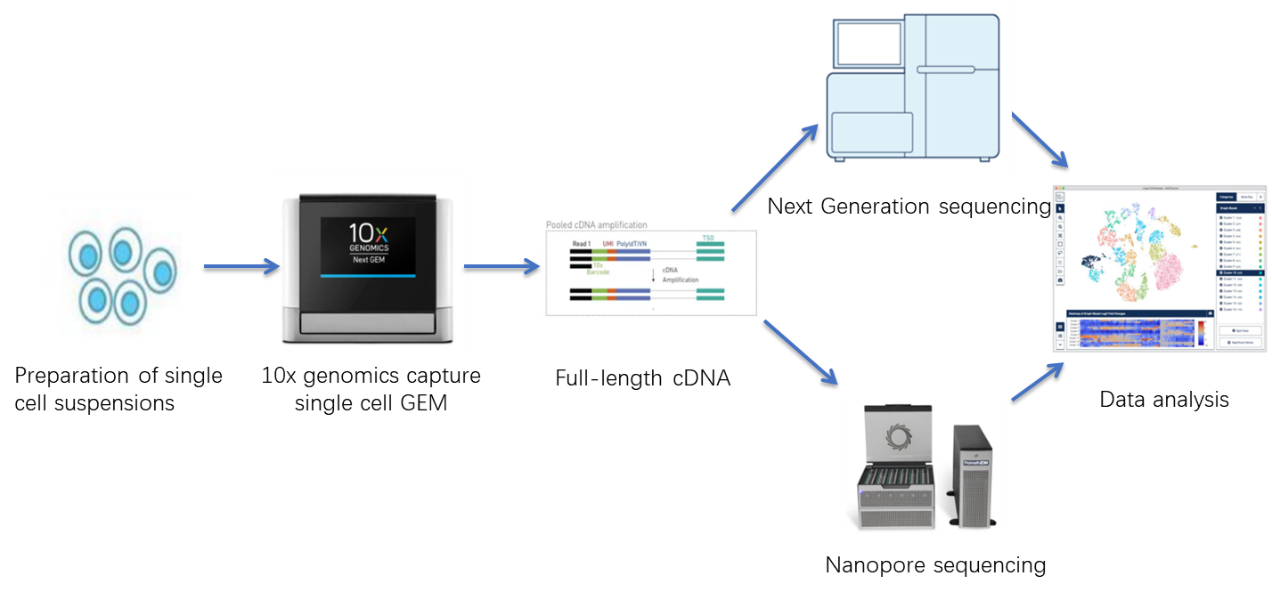
Function
Classification of cell subsets
Identification of rare cell types
Pathologic diagnosis
Screening of specific molecular markers
Application direction
Organ/tissue cell atlas
Developmental Biology Research
Disease-associated cell type Research
Tumor research
Virological research
-
-
FlsnRNA-seq: protoplasting-free full-length single-nucleus RNA profiling in plants
The broad application of single-cell RNA profiling in plants has been hindered by the prerequisite of protoplasting that requires digesting the cell walls from different types of plant tissues. Here, we present a protoplasting-free approach, flsnRNA-seq, for large-scale full-length RNA profiling at a single-nucleus level in plants using isolated nuclei. Combined with 10x Genomics and Nanopore long-read sequencing, we validate the robustness of this approach in Arabidopsis root cells and the developing endosperm. Sequencing results demonstrate that it allows for uncovering alternative splicing and polyadenylation-related RNA isoform information at the single-cell level, which facilitates characterizing cell identities.

Fig. 1 Schematic diagram of protoplasting-free single-nucleus RNA-seq.
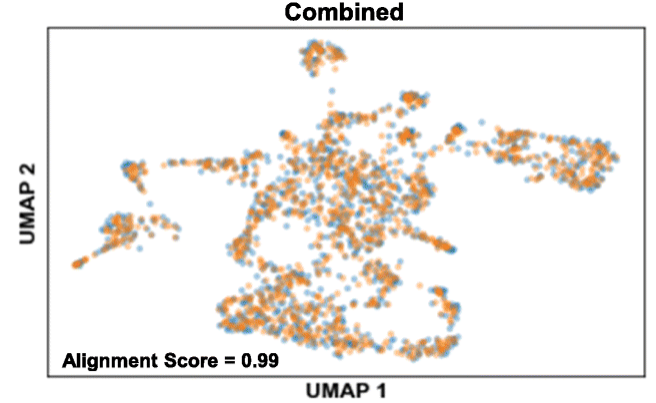
Fig. 2 The clustering result using Nanopore abundance matrix closely resembles the one generated by Illumina data.
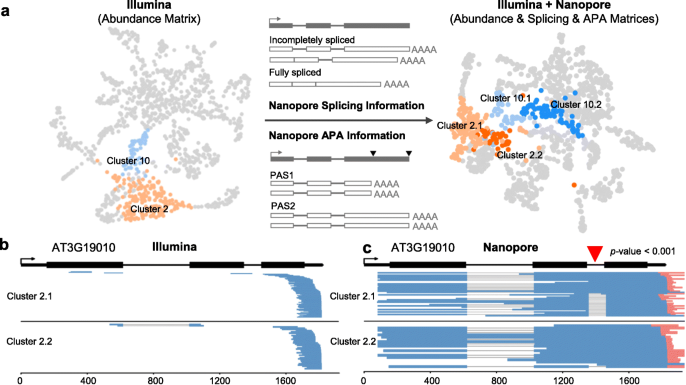
Fig. 4 We found that the original cluster 2 (mature non-hair) and cluster 10 (cortex) from Illumina data can be further separated into two subcell type clusters after the multilayer clustering. As an example, from the Illumina data, transcripts of AT3G19010 are present in both subcell type 2.1 and 2.2, while the Nanopore data revealed a large difference at the splicing level of this gene between the two sub-clusters, with the second intron largely unspliced in subcell type 2.2.
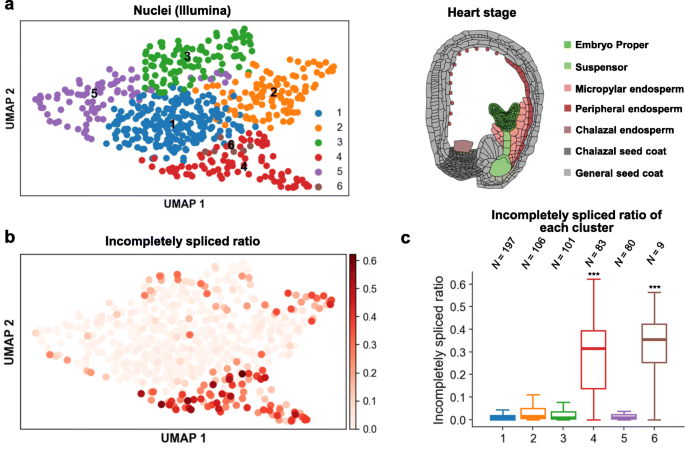
Fig. 5 We applied flsnRNA-seq to the multinucleate endosperm isolated from the heart-stage ovules of Arabidopsis and generated both the Illumina and Nanopore 10x libraries. We used Nanopore full-length transcript data to analyze retained introns in each nucleus and found that the nuclei from cluster 4, a major cluster that accounts for 14% of the total nuclei, exhibits a distinct high ratio of incompletely spliced transcripts.
REFERENCES
Yanping Long et al. FlsnRNA-seq: protoplasting-free full-length single-nucleus RNA profiling in plants .2021. Genome Biology.
-
Copyright © 2018 Wuhan Benagen Technology Co., Ltd . All Rights Reserved.



